

One of my favourites blogs, which I have mentioned in previous posts, is In the Pipeline, written by Derek Lowe. He posts several times per week (I wonder when he gets the time, maybe commuting), and sometimes it is a source of inspiration for our posts here. And the opposite: not the first time I have sent him materials for his blog. I have other favourite blogs, but they are hobby-related, so I will not mention them here, just in case (anyway, most of my acquaintances know that I am a bit of a freak).
A recent post in Lowe’s blog has triggered a reflection about how many times we have seen the phenomenon he highlights, probably most of them whithout even being aware of it. The post is related with a paper by Diederich et al. entitled Tracing Binding Modes in Hit-to-Lead Optimization: Chameleon-Like Poses of Aspartic Protease Inhibitors. Can you see where this is going?
In summary, the authors tried to develop a SAR series for a protease inhibitor. Having X-ray diffraction data of the protein-molecule complex and a computational model of the observed binding mode, they did the typical tweaks and changes in the inhibitor (see the figure below).
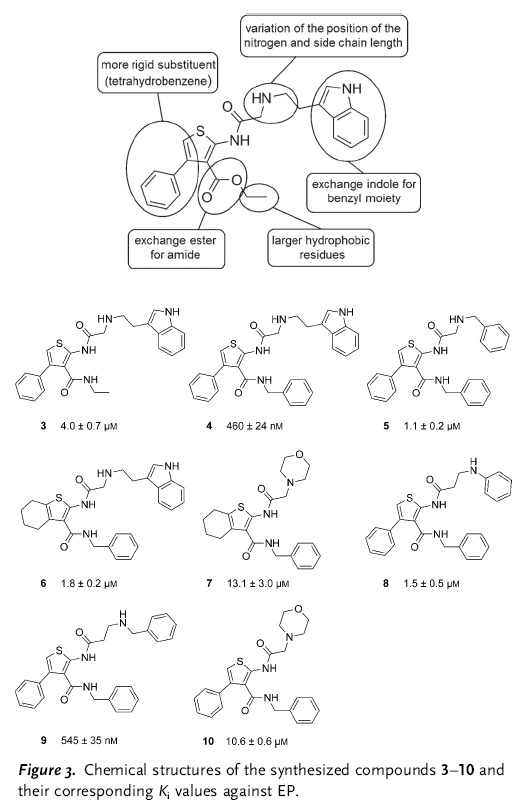
The results obtained for the SAR were very, very strange. For example, they changed the ester to amide to check the influence of the observed intramolecular H-bond (comp. 3). This change gave a five fold increase in activity. They anticipated a gain in affinity by replacing the ethyl ester with “larger hydrophobic substituents aimed to address the hydrophobic S2/S4 pocket” (comp. 4). Indeed, it boosted the activity by one order of magnitude. But “Removal of the 4-phenyl substituent and annulation of a tetrahydrobenzene moiety at the [b] site of the thiophene ring (6) reduces affinity by about fourfold, although […] this substituent appeared disordered and was not expected to contribute significantly”. They found the SAR “rather incongruent”, so much that they performed more structural analysis of the new molecules with the protease. And surprise, the binding poses of the analogs are different. So different, that in fact there are four different binding poses.
Maybe this is not common. Or maybe it happens often than we think. My own opinion: it happens often, but goes undetected, and we attribute the results to a myriad of different causes: lack of reproducibility in the primary assay; the compounds are not pure enough and the impurities are messing with the assay; the strange results are due to the presence of residual solvents… But I can easily think of three or four recent projects where those small changes in a chemical series gave so strange results that we were puzzled. Long conversations around the table, checking every bit of information, discussing how according to our previous experience that ring with a 2-chlorine or 4-chlorine should be not so different according to the model, or why a lipophilic pocket which should accept a bulkier group in fact does not, or why the expected H-bond in that moiety is not improving the binding. What was really happening there?
And at the end, usually, you drop the series, because it seems the SAR is not going anywhere. In discovery the time is so critical that usually you do not have the opportunity to dig for the explanation: you just keep going ahead, with other compounds, other chemical series. How many excellent opportunities have we lost by doing so?